4.6 Calculating the Reverse Complement in PerlAs you recall from Chapter 1, a DNA polymer is composed of nucleotides. Given the close relationship between the two strands of DNA in a double helix, it turns out that it's pretty straightforward to write a program that, given one strand, prints out the other. Such a calculation is an important part of many bioinformatics applications. For instance, when searching a database with some query DNA, it is common to automatically search for the reverse complement of the query as well, since you may have in hand the opposite strand of some known gene. Without further ado, here's Example 4-4, which uses a few new Perl features. As you'll see, it first tries one method, which fails, and then tries another method, which succeeds. Example 4-4. Calculating the reverse complement of a strand of DNA#!/usr/bin/perl -w # Calculating the reverse complement of a strand of DNA # The DNA $DNA = 'ACGGGAGGACGGGAAAATTACTACGGCATTAGC'; # Print the DNA onto the screen print "Here is the starting DNA:\n\n"; print "$DNA\n\n"; # Calculate the reverse complement # Warning: this attempt will fail! # # First, copy the DNA into new variable $revcom # (short for REVerse COMplement) # Notice that variable names can use lowercase letters like # "revcom" as well as uppercase like "DNA". In fact, # lowercase is more common. # # It doesn't matter if we first reverse the string and then # do the complementation; or if we first do the complementation # and then reverse the string. Same result each time. # So when we make the copy we'll do the reverse in the same statement. # $revcom = reverse $DNA; # # Next substitute all bases by their complements, # A->T, T->A, G->C, C->G # $revcom =~ s/A/T/g; $revcom =~ s/T/A/g; $revcom =~ s/G/C/g; $revcom =~ s/C/G/g; # Print the reverse complement DNA onto the screen print "Here is the reverse complement DNA:\n\n"; print "$revcom\n"; # # Oh-oh, that didn't work right! # Our reverse complement should have all the bases in it, since the # original DNA had all the bases--but ours only has A and G! # # Do you see why? # # The problem is that the first two substitute commands above change # all the A's to T's (so there are no A's) and then all the # T's to A's (so all the original A's and T's are all now A's). # Same thing happens to the G's and C's all turning into G's. # print "\nThat was a bad algorithm, and the reverse complement was wrong!\n"; print "Try again ... \n\n"; # Make a new copy of the DNA (see why we saved the original?) $revcom = reverse $DNA; # See the text for a discussion of tr/// $revcom =~ tr/ACGTacgt/TGCAtgca/; # Print the reverse complement DNA onto the screen print "Here is the reverse complement DNA:\n\n"; print "$revcom\n"; print "\nThis time it worked!\n\n"; exit; Here's what the output of Example 4-4 should look like on your screen: Here is the starting DNA: ACGGGAGGACGGGAAAATTACTACGGCATTAGC Here is the reverse complement DNA: GGAAAAGGGGAAGAAAAAAAGGGGAGGAGGGGA That was a bad algorithm, and the reverse complement was wrong! Try again ... Here is the reverse complement DNA: GCTAATGCCGTAGTAATTTTCCCGTCCTCCCGT This time it worked! You can check if two strands of DNA are reverse complements of each other by reading one left to right, and the other right to left, that is, by starting at different ends. Then compare each pair of bases as you read the two strands: they should always be paired C to G and A to T. Just by reading in a few characters from the starting DNA and the reverse complement DNA from the first attempt, you'll see the that first attempt at calculating the reverse complement failed. It was a bad algorithm. This is a taste of what you'll sometimes experience as you program. You'll write a program to accomplish a job and then find it didn't work as you expected. In this case, we used parts of the language we already knew and tried to stretch them to handle a new problem. Only they weren't quite up to the job. What went wrong? You'll find that this kind of experience becomes familiar: you write some code, and it doesn't work! So you either fix the syntax (that's usually the easy part and can be done from the clues the error messages provide), or you think about the problem some more, find why the program failed, and then try to devise a new and successful way. Often this requires browsing the language documentation, looking for the details of how the language works and hoping to find a feature that fixes the problem. If it can be solved on a computer, you can solve it using Perl. The trick is, how exactly? In Example 4-4, the first attempt to calculate the reverse complement failed. Each base in the string was translated as a whole, using four substitutions in a global fashion. Another way is needed. You could march though the DNA left to right, look at each base one at a time, make the change to the complement, and then look at the next base in the DNA, marching on to the end of the string. Then just reverse the string, and you're done. In fact, this is a perfectly good method, and it's not hard to do in Perl, although it requires some parts of the language not found until Chapter 5. However, in this case, the tr operatorwhich stands for transliterate or translationis exactly suited for this task. It looks like the substitute command, with the three forward slashes separating the different parts. tr does exactly what's needed; it translates a set of characters into new characters, all at once. Figure 4-2 shows how it works: the set of characters to be translated are between the first two forward slashes. The set of characters that replaces the originals are between the second and third forward slashes. Each character in the first set is translated into the character at the same position in the second set. For instance, in Example 4-4, C is the second character in the first set, so it's translated into the second character of the second set, namely, G. Finally, since DNA sequence data can use upper- or lowercase letters (even though in this program the DNA is in uppercase only), both cases are included in the tr statement in Example 4-4. Figure 4-2. The tr statement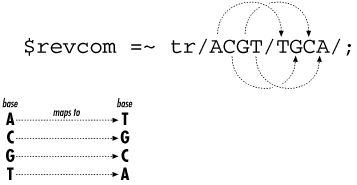 The reverse function also does exactly what's needed, with a minimum of fuss. It's designed to reverse the order of elements, including strings as seen in Example 4-4.
|
Index terms contained in this sectionDNAreverse complement of strands, calculating Perl reverse complement, calculating reverse complements calculating in Perl strands of DNA, calculating reverse complement |